












-
格列卫传奇:磨难,坚韧,浴火重生
元沙优投 / 2018-07-07 11:47 发布
原创: Jerry Pharmcube

To Dr. Brian J. Druker
2000年2月的一天,Lopossa从奥林匹亚一家医院的病床上醒来。她拉开窗帘,看着窗外的风景,“就这样子结束吧”,她这样想。
但仍有太多的东西让她割舍不下,她的丈夫,她的孩子,她的孙子孙女,她的那些朋友。可即使割舍不下又能怎样呢,身体的疼痛已经让她无法承受。已经到了告别的时候了。
她的面色苍白,血液中的红细胞已经少得可怜,她变得虚弱无力,行走都已经非常困难。满是白细胞的脾脏已经被撑得如同甜瓜般大小,她的呼吸也开始变得困难,她时常呕吐,病魔无时无刻不在折磨着她的肉体。
她的骨头深处传来阵阵刺痛,她的骨髓正在疯狂地制造着白细胞。发烧,冷颤,无法言说的寒冷。裹着医院的毯子,她的身体仍然如同掉进了冰窖。
Lopossa患有慢性粒细胞白血病(CML),由于年龄太大,病情过于严重,她已经无法进行骨髓移植。她尝试过很多药物,干扰素也是其中之一。但干扰素令她的恶心呕吐更加严重,使她的疼痛变得更加难以忍受。
这些让人恐惧的副作用最终使她决定放弃药物治疗。Lopossa的丈夫George把她接回家中。她的孩子们也纷纷从外地赶来,和她道别。
Lopossa连自己葬礼上的音乐都选好了,其中的一首是Because I Have Been Given Much, 她希望她的孙子孙女能在她的葬礼上演唱这首歌。
但Lopossa还有一件事情未完成。在她与家人进行道别之后不久,她勉强支撑着,抽出了几天时间,前往俄勒冈卫生科学大学(OHSU)赴约。
1999年12月初的一天,George开车来奥林匹亚医院探望Lopossa。他把车停在一家超市边上,顺手买了一张报纸。而这份报纸上刊登的一条新闻却吸引了他的注意。那是关于一款CML药物的报道,该药物正在OHSU进行临床研究。
Lopossa的医生设法让她加入了该药物的II期临床试验。虽然早已与家人进行过告别,但接下来的几个月里,这款药物,却让她重获新生。
费城故事
David Hungerford不敢相信自己的眼睛。他凑到显微镜前看了又看,没有错,其中的一条染色体变短了。
那还是1959年,遗传学领域才刚刚起步。也仅仅是在三年前,科学家才确定人细胞内染色体的正常数目是46条。
Hungerford在费城癌症研究中心工作,他曾夜以继日地观察果蝇的染色体,这使他对染色体的结构变得格外敏感。
上世纪五十年代,遗传学的学术圈还很小。整个费城里,对遗传学感兴趣的科学家一只手也能数得的过来。因此Hungerford与Peter Nowell的合作也算是水到渠成。
1956年Nowell从海军退役,回到了他的故乡费城,在宾夕法尼亚大学从事癌症研究。Nowell一直在使用一种很有意思的方式来观察染色体。与以往操作不同,他将细胞用水涨开,然后再用染料对细胞进行染色。
而正是这种操作帮助了Hungerford更好地看清细胞内的染色体。1959年,也就是在他们相遇的两三年之后,他们发现CML患者癌细胞内的一条染色体存在异常。
Hungerford觉得很诧异,他按下相机的快门,拍下了一张足以改写肿瘤领域,甚至整个医学领域的一张照片。
Nowell 以及 Hungerford将这项研究于1960年在Science上发表。当时的人们并不会意识到,这短短的三百字,将预示着肿瘤治疗新时代的开启。
在Nowell 和Hungerford发表第三篇论文,报道他们在更多的CML病人癌细胞内发现这种异常的染色体时,这条染色体被正式命名为费城染色体,以纪念它被发现的城市。
芝加哥往事
尽管费城染色体与CML之间存在着某种关联,当时也很少有人去怀疑染色体异常与癌症的形成是否存在因果关系。
究竟是白血病导致染色体缩短,还是染色体异常导致白血病的形成?
尽管后来人们发现费城染色体是在22号染色体的两条拷贝中的一条出现了异常,但是由于当时技术的限制,没有人能够进行进一步研究,也没有人能够给出答案。
而费城染色体在十年的沉寂之后,终于迎来了爆发。
而这次爆发背后的驱动因素,正是技术的进步。直到1969年,Giemsa染液染色仍然是观察染色体的最好方式。但Giemsa染液只能够将染色体染成统一的颜色,这也极大限制了Nowell和Hungerford当时的研究。
但在1970年,Janet Rowley听说当时已经有人能够以一种全新的方式来观察染色体。
Rowley是芝加哥大学的遗传学家,她对染色体领域的研究历史有着很深入的了解,而且也非常想进行基因与癌症关联的研究。她当时了解到,英国已经有人能够使用这种新型的染色方法来观察染色体。
1971年,恰逢Rowley的学术休假(sabbatical),她决定与丈夫前往牛津大学学习这项技术。虽然这项技术的操作相对复杂,但确实会取得意想不到的效果:在Giemsa 染料染色之前,需要使用一种强大的荧光染料quinacrine mustard对细胞进行预处理。之后在荧光显微镜下,染色体就不会是单色的了,而是呈现出黄绿相间的条纹。
在回到芝加哥之前,Rowley已经熟练地掌握了这项技术。所以当她回到了芝加哥,她就开始尝试使用这项技术来观察CML病人的费城染色体。
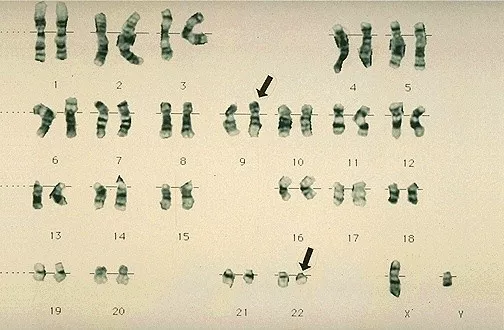
他观察到,在所有的样本中其中一条22号染色体都会存在异常。但是在1972年的时候,她察觉到了另一项怪事。她发现样本中的9号染色体也存在异常。样本中9号染色体比正常细胞的9号染色体要长。
这种差别并不是很大,如果没有这项新技术,她大概永远也不会观察到这一现象。
她同时也发现,22号费城染色体的缺失,与9号染色体的异常永远是同时存在的。而且似乎22号染色体缺失的部分正是9号染色体增加的部分。她当时并不确定,但她知道这两条染色体之间一定存在某种关联。
到底22号染色体与9号染色体存在什么关联,这与CML的形成又有什么关系?
基因融合
长久以来,两位荷兰的遗传学家Nora Heisterkamp 以及 John Groffen都想克隆染色体。但一开始他们并没有想去专门研究CML。
1978年,Heisterkamp 和 Groffen前往美国国家癌症研究所的Baltimore实验室进行技术学习。Groffen之前就读过Baltimore实验室的Abl的相关文章,Groffen对Abl很感兴趣,希望能够找到Abl基因所在的染色体位置。
为此Groffen设计了一种Abl探针,以此来寻找Abl的位置。1982年,他找到了答案,人abl基因存在于9号染色体上。
一天傍晚,Heisterkamp 和Groffen俩人凑到一起喝酒,期间他们讨论起了Abl。Abl存在于9号染色体上,是不是与CML会有某些关联?
很凑巧,Heisterkamp有一个表兄弟也是遗传学家,而他最近在克隆费城染色体的断裂位点。这真是绝妙的机会。
在这之后,Heisterkamp以及Groffen将他们的abl探针寄给了这位在荷兰的遗传学家Gerard Grosveld,让他使用这个探针来寻找与之匹配的费城染色体位点。
很快他便找到了探针配对的基因位点。但是令他诧异的是,匹配位点并不完全存在于两条9号染色体。他发现其中一条9号染色体存在abl,而另一条9号染色体中,abl却存在异常,该基因的一大部分缺失,而缺失的部分,却存在于22号染色体上。
我想大家很容易就能够体会到这项发现的重要性。这一发现证明了Groffen以及Heisterkamp之前的推测是正确的。
那么接下来的问题是,abl转移之后到底与什么基因相连呢?
Heisterkamp 以及Groffen发现,9号染色体转移的片段转移到22号染色体上之后,与bcr基因相连。
在含有费城染色体的CML细胞中,abl与bcr产生了融合。这两段之前并无相关性的基因序列现在融合在了一起,成为了bcr/abl。
而接下来,最重要的问题是,bcr/abl是如何导致癌症的形成的呢?
部分证据来源于K562细胞,K562细胞来源于CML病人。1984年,Witte发现这些细胞内的激酶活性能够持续性的处于活化状态。两年之后,Baltimore实验室发现bcr/abl与这种激酶的持续活化状态相关。
虽然没有更为直接的证据,但是此时,已经有很多人无比确信bcr/abl能够导致CML。
而这其中的一位,正是Brian Druker。
靶向治疗的诞生
上世纪八十年代末,Druker陆续的听到有人谈论通过设计小分子药物,来抑制酪氨酸激酶的活性,从而治疗癌症。
如果说某些癌症是由酪氨酸激酶活性异常导致的话,那么抑制激酶活性应该能够达到治疗癌症的目的。
但在那时,Druker才刚开始学习基本的实验室技能。而对于当时的Nick Lydon来说,他却已经对激酶有了非常深入的了解,而且他也已经准备好进行激酶抑制剂的研究。除了Lydon,他的老板Alex Matter也对激酶抑制剂满怀信心。
上世纪八十年代早期,Matter从Schering离职,前往瑞士的一家公司Ciba-Geigy。尽管前一份工作并不顺心,但他之前在Schering的时候就已经对癌症研究有着非常浓厚的兴趣,
当Matter还在Schering的时候,他就经常与Lydon讨论激酶。在Matter来到Ciba-Geigy之后,激酶与原癌基因的关联研究也已经越来越多。
Matter和Lydon当时自然也了解到了Bcr/Abl酪氨酸激酶与费城染色体的关联,几乎所有的CML病人都存在这种突变。
虽然当时依然没有十足的证据,但在众多关于激酶与癌症关联的研究中,Bcr/Abl是研究最清楚的一个。
来到Ciba-Geigy之后, Matter希望开始进行激酶抑制剂的研究,Matter以及Lydon都认为激酶会是非常好的治疗癌症的靶点。
Lydon之前已经有了数年的激酶研究经验。1984年夏天,Lydon接到了Matter打来的电话,邀请他来Ciba-Geigy,加入激酶抑制剂的项目。
公司当时同意资助这个项目,但由于之前在肿瘤领域受挫,公司并不愿意在这种看似风险极大的项目上投入过多的资金和精力。
但Matter的老板,同时也是他的好友,告诉Matter他能够进行肿瘤药物的研发项目。但是他同时也被告知老板不能保证公司在这个项目上有很大的投入。因此除了激酶抑制剂,Matter同时也在做其他药物研发项目。而激酶抑制剂只能在悄无声息的情况下进行,当时也并没有引起多少人的注意。
激酶抑制剂的药物研发与传统肿瘤药的研发存在着很大的区别,Ciba-Geigy进行的激酶抑制剂研发应该是药物研发史上最早期的激酶抑制剂研发项目之一。
通过设计靶向单一(或几个)靶标的药物来治疗癌症,这种策略叫做理性药物设计。在CML的例子中,由于bcr/abl很可能是导致CML的根源,因此通过设计药物来阻断相关激酶的活性,理论上来讲能够达到很好的治疗癌症的目的。
虽然理性药物设计的原理很简单,但实现起来确实极其困难。当时设计激酶抑制剂的目的是通过小分子抑制剂与激酶结合,阻断ATP与激酶的结合过程。
虽然不同激酶的ATP结合腔存在些许差异,但这种差异并不大。如果药物同时抑制过多种类的激酶,将有可能产生严重毒性,导致病人死亡。而克服这种选择性问题,也是激酶抑制剂药物研究的关键所在。
在Lydon和Matter进行激酶抑制剂研究的八十年代中期,已经有很多实验室报道了多种化合物能够产生激酶抑制作用,但无一例外,这些化合物的选择性都非常差。
除此之外,化合物的活性筛选过程也是问题重重,如果没有一个良好的药物活性评价体系,设计和合成再多的化合物又有什么用呢?
然而,Lydon和Matter最大的困境并不是来源于科学研究,而是当时公司对于该研究项目的态度。
尽管当时还没有找到bcr/abl能够CML导致的确凿证据,但他们始终相信bcr/abl激酶抑制剂的价值。
但即使bcr/ab与CML存在非常强的相关性,CML的研究对于制药公司来说也很难产生足够的吸引力。问题在于CML的发病率并不高,美国每年的新发病人数量只有5000人,全球总体的发病率只有大约十万分之一。
在当时来说,这样的病人群体很难对制药公司产生吸引力,治疗更常见的癌种的药物会带来更大的利润。
而这也是为何当时PKC, PDGFR以及EGFR相比Abl能够吸引更多关注的原因,因为这些激酶能够在多种类型的常见肿瘤中显现相关性。
但Lydon和Matter依然坚持,因为他们认为CML与Abl的相关性是当时发现的与癌症相关性研究中关联最强的一种激酶。
上世纪八十年代末,Druker 与Lydon 的联系也变得越来越频繁。他们都相信Bcr/Abl酪氨酸激酶能够导致CML,尽管确凿的证据在1990年才出现。
在科学探索方面,尽管对于疾病机制有着十足的信心,Lydon和Matter团队筛选了一个又一个的化合物,但这些化合物的实验都失败了,至少没有一个化合物能满足激酶抑制的特异性的要求。
但最终,他们经过一系列的结构设计与修饰,找到了一个选择性高,活性好的化合物。而这个化合物也正是伊马替尼。
时间到了1993年。当时Ciba-Geigy处于领先地位的激酶抑制剂项目并不是Abl激酶抑制剂,而是另一款药物,一款靶向PDGFR激酶的抑制剂。
尽管Lydon始终相信Abl激酶抑制剂治疗CML的潜力,但CML的低发病率,有限的市场空间仍然不会让公司的管理层产生什么兴趣。
Lydon一直要求对伊马替尼进行进一步的研究,他相信这款药物的潜力,他知道病人在等待着新药物的上市,他相信这是一款能够拯救CML患者生命的药物。
同样焦急等待着的,还有Druker。何时进行毒理学评价?何时提交IND?何时药物才能够进入临床研究?
焦急等待中的Druker决定进行另一种策略,以证明伊马替尼治疗CML的潜力:将伊马替尼与患者的骨髓细胞混合,并检测伊马替尼是否能够抑制CML细胞。这项研究的成功至少证明了伊马替尼能够有效地杀死人的CML细胞,而对正常人体细胞没有很大影响。
有了这项研究,Lydon以及Matter就有了更加有力的证据说服管理层推进伊马替尼的临床前研究。
最终在1995年,公司同意进行伊马替尼的临床前的研究。而在接下来的毒理学研究中,需要证明伊马替尼的在动物模型中的安全性。而正是这些研究,几乎将伊马替尼的前途断送。
祸不单行
Ciba-Geigy决定将伊马替尼做成注射剂之后进行毒理学研究。1996年的7月,他们终于拿到了第一批毒理学实验的结果。结果发现,以静脉形式接受给药的狗会在导管末端出现血凝块。
该研究的失败让公司内部对于伊马替尼的信心大减。但Matter认为,如果静脉注射不成功,那么口服制剂是否能够避免毒性的产生呢?Matter告诉Druker公司之所以决定做注射剂,是因为他们错误的预计伊马替尼做成口服制剂无法有效吸收。
然而如果做成口服制剂,毒理学评价又得从头开始。也就是说,伊马替尼能进入临床研究的最早时间也只能是在1997年的三月。
除了注射剂毒性风波的影响,接下来发生的事情,也让伊马替尼的研究项目遭受了极大的威胁。
就在伊马替尼注射剂进行毒理学评价的时候,Ciba-Geigy与Sandoz合并了。而这个合并之后的公司,正是诺华。
当诺华成立的时候,之前的很多研究项目都面临着被中止的困境。当时的诺华是不会有兴趣去开发一种市场空间不大,而且存在毒性问题的药物,试图去将其推向临床研究。
Lydon屡次受挫,在诺华成立之后他便辞职了。
但毒理学研究仍在持续,很长一段时间内,Druker都没有从诺华那边得到什么消息。最终,消息从诺华传来,仍然是坏消息。
600mg剂量的伊马替尼能够引起狗的肝衰竭。在大鼠的研究中,更低剂量的药物也同样能够引起肝损伤。
这一次出现的毒性问题同样引起了不小的震动。但Druker相信,是给药方式存在问题,而且应用的剂量比临床中应该使用的剂量要高得多。如果在高剂量给药时出现转氨酶异常之后及时停止用药,就不会出现什么大的问题。
但诺华却不这么认为。就在此时,第七项毒理学实验开始了,在猴子的实验中,他们并没有发现毒性问题。
与此同时,Druker直接与FDA取得了联系,告诉他们诺华已经有了足够的安全性数据提供给FDA进行审查。而FDA也认为他们已经有了足够的安全性数据。
但当Druker把消息告诉诺华的高管之后,他们仍然不为所动。
如果他们连FDA的建议都不采纳,还能有什么办法说服他们进行临床实验呢?
Lydon当时给Druker提供了一个建议。跟诺华的高管协商,如果不愿意进行临床试验的话,可以将该项目出售。由于已经有了非常充足的前期数据,这个项目对于很多制药公司来说是非常有吸引了的。Lydon也对Druker说,他自己成立的公司很愿意收购这个项目。卖掉也总比将伊马替尼束之高阁来的好。
最终,诺华还是同意了尝试进行临床试验。
从激酶抑制剂项目启动的1984年,到1990年先导化合物筛选,到1998年的一期临床实验,伊马替尼经历了无数的波折才开始绽放它的光芒。
浴火重生
一条陡峭蜿蜒栽满行道树的道路通往OHSU的主校区,这里位于马奎阿姆山的峰顶附近。一到雾天,整个校园就像是童话里的仙境一般。
通过空中缆车也可以去往OHSU校园,敞车沿着五号州际公路上空的钢索呼啸而过,在威拉米特河和一家医院的站台之间来来回回地运送着游客。
2000年2月,Lopossa前往OHSU赴约。Lopossa参加了伊马替尼的II期临床试验,当时全世界范围内十二家医疗中心的约五百名病人参与了这项试验。
她当时被George搀扶着挪进了诊所。真的像是无药可救了吧。她的白血球计数已经超过了20万,比正常值高出二十多倍。早已经没有别的治疗选择了。
Druker团队给Lopossa做了检查,并给她服用了一粒伊马替尼,但她却没法咽下去。
第二天早晨,George和Lopossa在她妹妹的公寓中醒了过来,George给她做了一份奶昔。终于,那粒伊马替尼被咽了下去。第二天照旧,就这样一直坚持了下去。
三周之后,她的脾脏恢复到了正常状态,Druker说,她自我感觉很好。白血球计数也降了下来。在濒临死亡之后绝地重生,这真的是一个奇迹。2000年五月份,Lopossa去她母亲的墓地扫墓,旁边是她之前为自己准备的墓地。“我本该已经长眠于此,”她对丈夫说。
“你现在不是还活蹦乱跳的吗,干嘛不拍张照片?”

2001年5月,伊马替尼 (格列卫) 获得FDA批准上市。

 公安备案号 51010802001128号
公安备案号 51010802001128号